渤健-临床候选药物布鲁顿酪氨酸激酶抑制剂-BI
Bruton的酪氨酸激酶(BTK)是蛋白质酪氨酸激酶(TFK)Tec家族的成员,该家族在B细胞中抗原受体(BCR)和中性粒细胞和嗜碱性粒细胞等髓样细胞Fc受体(FcR)中起作用。BTK激活BCR信号通路,驱使磷脂酶C-γ(PLCγ),磷脂酰肌醇3-激酶/ Akt和NF-κB的激活,最终调节B细胞激活和免疫球蛋白(Ig)的表达。
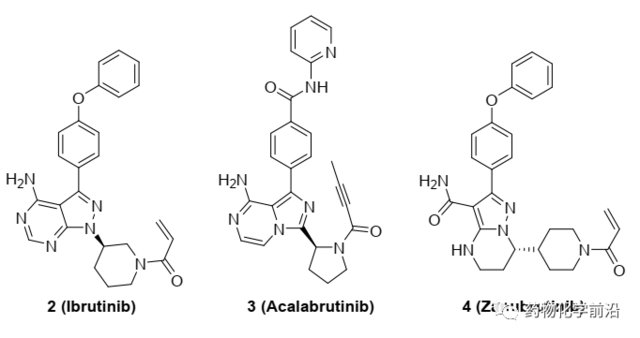
依鲁替尼是第一代共价BTK抑制剂,于2013年被FDA批准用于治疗患有套细胞淋巴瘤(MCL)和慢性淋巴细胞性白血病(CLL)的患者。第二代Acalabrutinib和Zanubrutinib是选择性更好的共价BTK抑制剂。现有的共价抑制剂虽然临床效果显著,但这些共价BTK抑制剂会导致肝氨基转移酶水平升高,在各种淋巴瘤中均会产生获得性耐药。而可逆的选择性BTK抑制剂的开发可以为自身免疫性患者提供相同的治疗效果,并降低风险。Fenebrutinib作为新一代的可逆抑制剂,正在进行类风湿性关节炎二期临床研究,这也说明可逆抑制剂在自身免疫性疾病的前景。
Hit化合物5是以前报道的BTK抑制剂,其激酶活性为3700nM,晶体结构结果表明,其结合模式与依鲁替尼是相反,其苯甲酰胺结构能够靶向BTK结构中的H3口袋,而这一口袋是实现BTK与其他Tec家族选择性的重要结构。
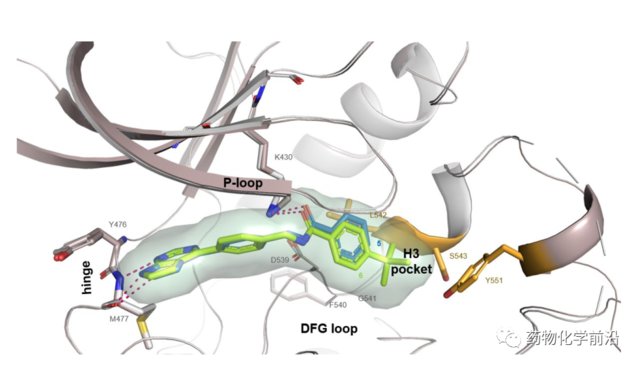
对晶体结构的进一步研究发现,H3口袋空间位置大,于是作者引入了空间位阻较大的叔丁基,得到化合物6,发现化合物6的活性提升了20倍,进一步对化合物6的晶体结构研究表明,苯环上2-取代可以与富含甘氨酸的P发生环疏水相互作用。

于是作者在苯环2位引入了一系列取代基,发现给电子取代基甲基能够显著提升约30倍的活性。

虽然化合物7在激酶活性上有了显著的提升,但是其成药性仍然较差,于是为了提高水溶性,降低清除率,以及提高WB细胞活性,作者在此基础上进行进一步的修饰。
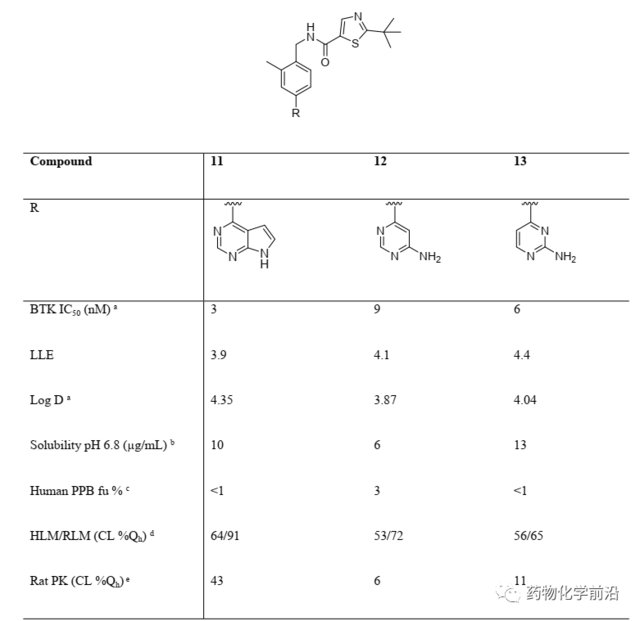
优化得到的化合物12和13在降低清除率的同时也保持了激酶活性,证明了这一修饰的正确性,但是在研究中发现,化合物13在WB细胞上的活性都在微摩尔级别,与激酶活性不匹配,于是作者继续进行修饰。
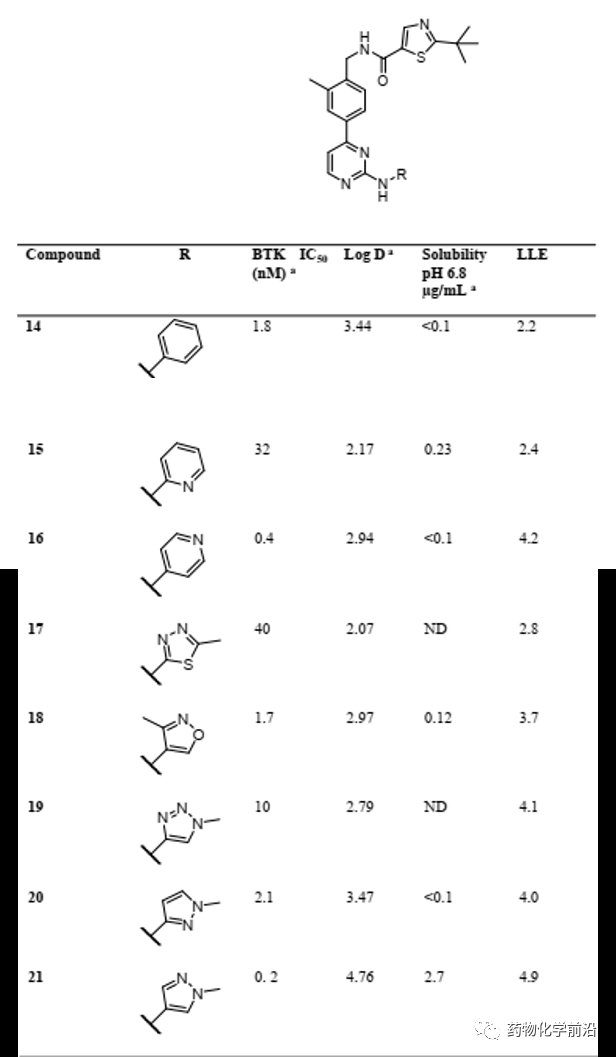
进一步优化得到化合物21,虽然活性有了显著的提升,但是其成药性仍然不好,于是作者将修饰位置着眼于靶向H3口袋的苯甲酰部位,电子建模研究表明,H3口袋中的不饱和芳香环能够被SP3杂化的取代基替代,因此作者在该位置引入了饱和烷烃杂环,以期能够提升化合物的水溶性。

这次的优化得到了化合物1,化合物1大大降低了亲脂性(LogD = 2.32),提高了溶解度,降低了血浆蛋白结合,并且提高了WB细胞的活性,此外其成药性较好(LLE = 5),在大鼠中的口服生物利用度为48%。进一步的晶体结构证实了其2-氨基吡啶亚胺部分起着双氢键受体/供体的作用,其残基位于蛋白质的铰链区。苯环上的甲基取代基位于P环下方,苄基脲接头将氮杂环丁烷伸到H3口袋中。

进一步对化合物的激酶选择性研究表明其激酶组最低选择性为433倍。
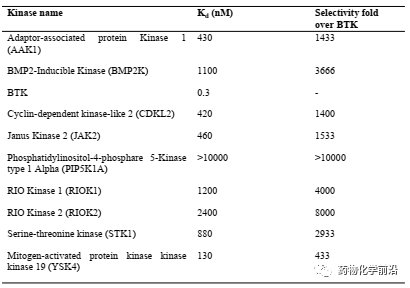
与依鲁替尼的泛抑制相比